
在美國布朗大學的一項突破性研究中,研究人員在藥物發現和理解蛋白質的複雜活動方面取得了關鍵的飛躍。這一進步,通過創新使用AlphaFold 2來利用人工智能的力量,爲預測蛋白質的動態構象(dynamic conformations of proteins)設定了新的前沿。這項研究發表在《自然通訊》期刊上,不僅推動了我們對蛋白質動力學的理解,而且有望加快新療法的發展。
揭開蛋白質之舞的面紗
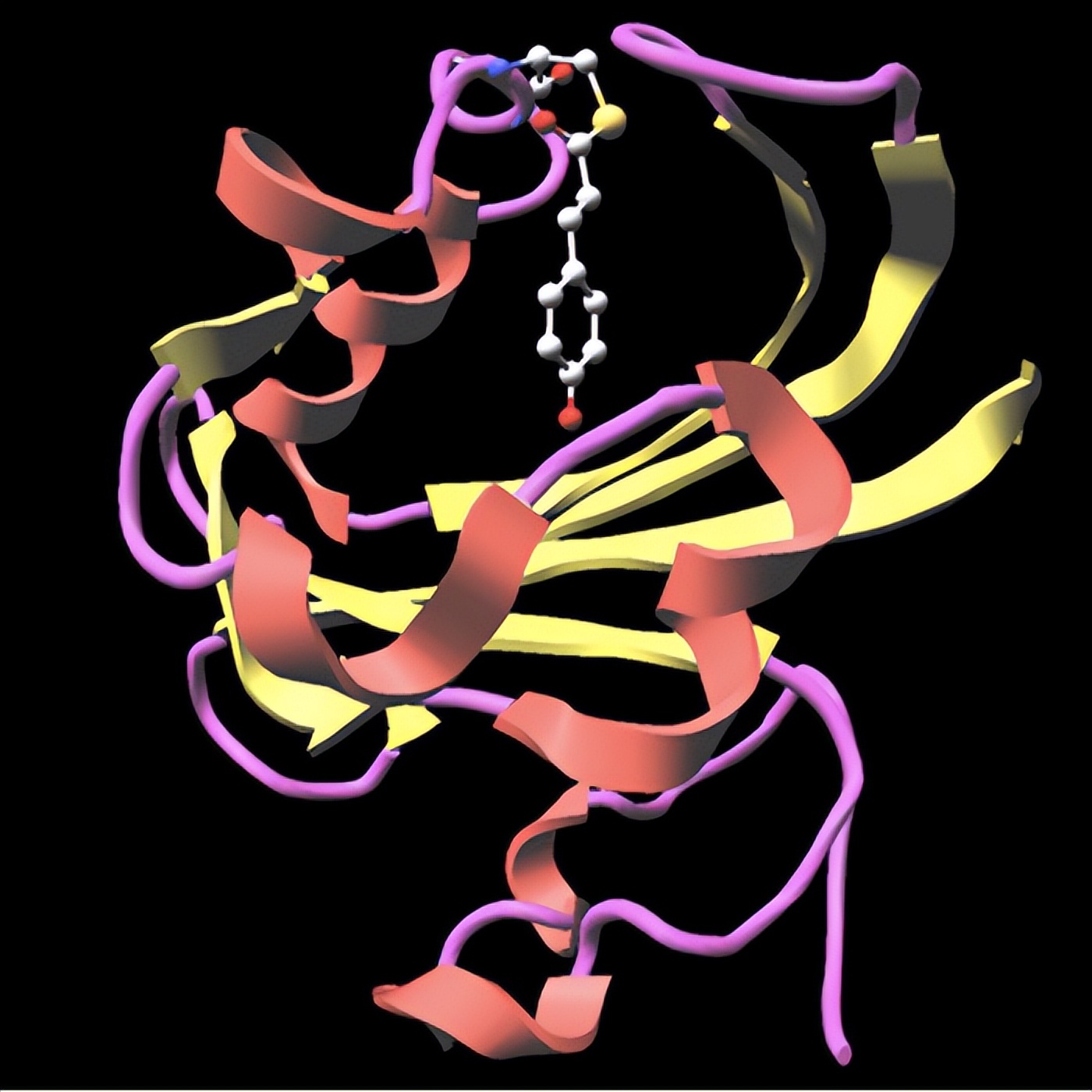
蛋白質及其複雜的結構,幾乎是每個生物過程的核心。了解這些分子如何折疊和改變形狀是一個重大的科學挑戰,因爲這是一個與其功能內在相關的過程。傳統上,X射線晶體學和核磁共振 (NMR)光譜等工具已被用于破譯蛋白質結構。然而,這些方法往往未能捕捉到蛋白質的動態性質,因爲它們在生物功能中變形和彎曲。

AlphaFold 2模型是由DeepMind開發的人工智能驅動的工具,它已經通過准確預測蛋白質結構引起了廣泛關注。但布朗大學的團隊並沒有止步于此。他們將AlphaFold 2的能力擴展到靜態預測之外,使其能夠對蛋白質潛在形狀的全系列(the full spectrum of a protein's potential shapes.)進行建模。這一發展標志著向前邁出的關鍵一步,類 似于捕捉蛋白質運動的高分辨率視頻,而不是單一的快照。
用機器學習彌合差距
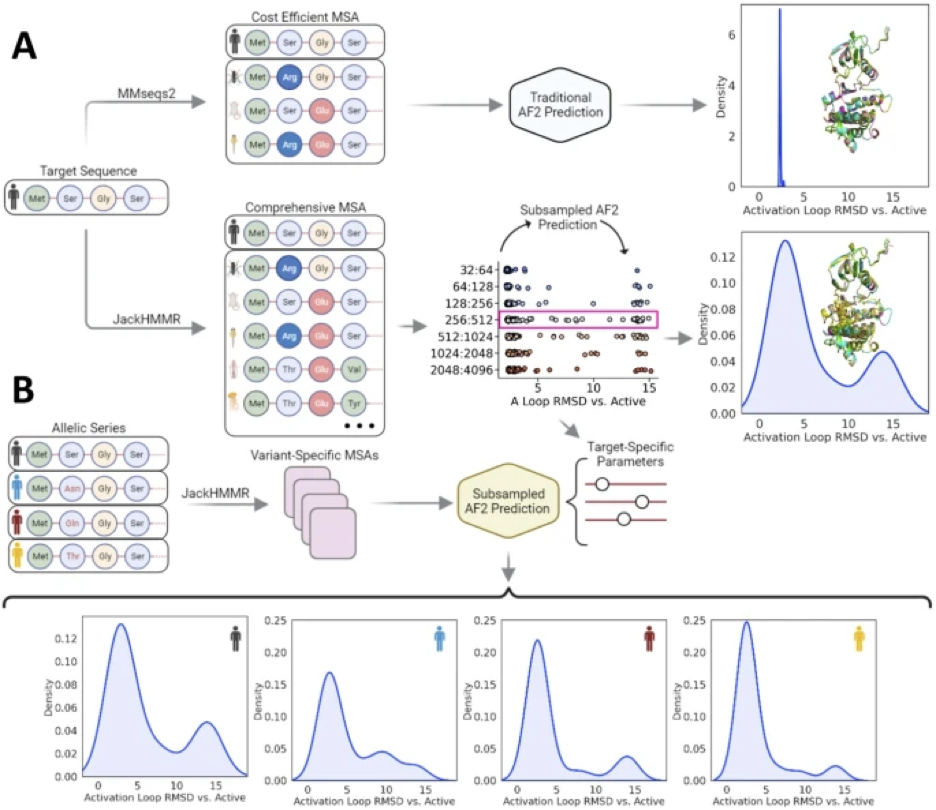
這一突破的關鍵在于一種名爲子采樣多序列對齊( subsampling multiple sequence alignments, MSA)的巧妙方法。通過選擇性地向AlphaFold 2提供已知蛋白質序列的大量數據庫的一部分n內容,研究人員告訴人工智能模型不僅預測一種,而且預測蛋白質可以假設的多種配置。根據核磁共振實驗的黃金標准驗證,這種方法展示了超過80%的令人印象深刻的准確率,這一壯舉充分說明了其潛力。
這種方法的與衆不同之處在于其普遍性和效率。在兩種蛋白質(Abl1激酶和粒細胞-大噬細胞菌落刺激因子,Abl1 kinase and granulocyte-macrophage colony-stimulating factor(GMCSF))上進行了測試,無論可用序列數據量如何,該方法都被證明是成功的。這種多功能性凸顯了其在各種蛋白質中的應用潛力,爲藥物發現開辟了新的途徑,特別是在靶向癌症治療領域。
從靜態到動態:蛋白質建模的新維度

蛋白質從靜態建模到動態建模的飛躍不僅僅是技術上的勝利;它代表了科學家如何進行藥物發現的範式轉變。傳統上對蛋白質基態結構的關注限制了研究人員了解蛋白質功能的全部範圍的能力,進而廣之,藥物如何有效地靶向這些分子。通過闡明蛋白質的構象景觀,這項研究爲設計根據其目標的動態性質進行微調的藥物鋪平了道路。
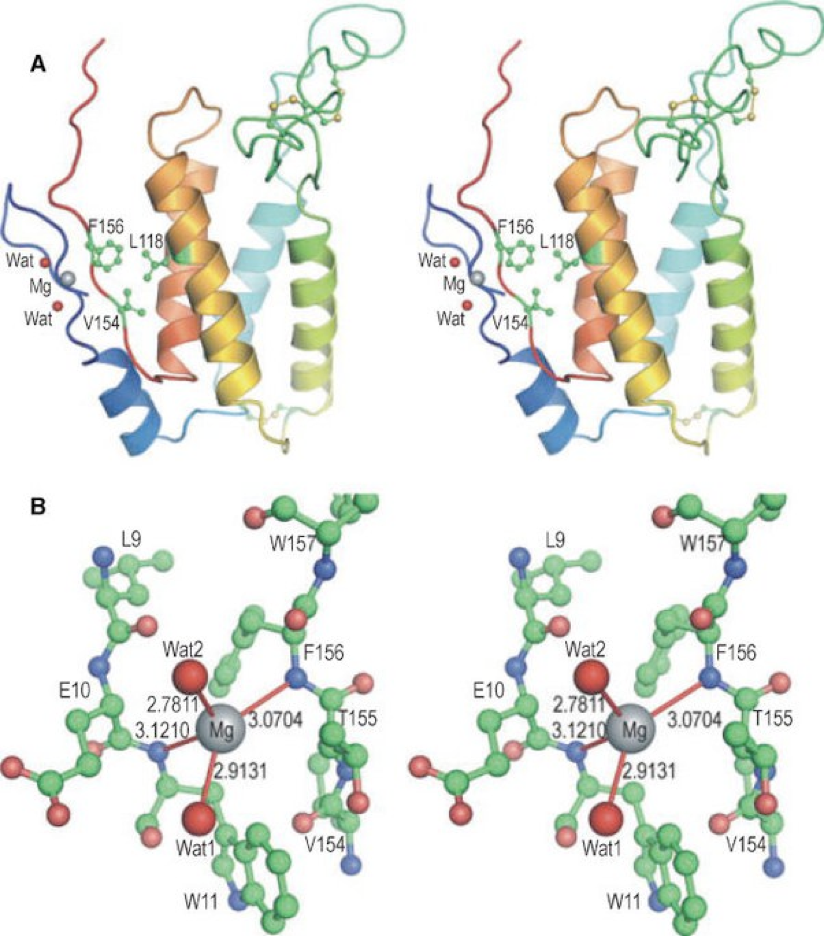
這項研究不僅擴展了我們的科學工具包;他們從根本上豐富了我們對生物過程的理解。他們的工作強調了一個關鍵的洞察力:要真正掌握藥物如何與蛋白質相互作用,必須考慮蛋白質可以采用的各種形狀。
影響和未來方向
這項研究的影響是廣泛而多樣的。例如,它提供了一個新的視角,通過它來看待耐藥性的挑戰——這是治療癌症等疾病的一個重要障礙。通過繪制蛋白質結構不斷變化的景觀,研究人員可以預測突變如何改變藥物的功效,指導對耐藥性更具彈性的下一代療法的開發。
此外,這種方法證明了跨學科合作的力量,融合了計算生物學、化學和機器學習領域,以應對醫學中一些最緊迫的挑戰。它還強調了人工智能和機器學習在科學發現中發揮的日益關鍵的作用,提供了前所未有的預測能力和效率的工具。

展望未來,布朗大學的團隊專注于完善他們的技術,提高其准確性,並擴展其適用性。隨著這項研究的不斷發展,它有望解鎖新的治療目標,揭示蛋白質動力學的奧秘,並最終迎來藥物發現的新時代。
